Entretien avec Sandipan Mohanty du Jülich Supercomputing Centre
Physicien et spécialiste du codage, Sandipan Mohanty travaille depuis 20 ans sur des simulations de biologie moléculaire pour les superordinateurs les plus rapides du monde. Ils permettent de décrypter les éléments constitutifs du vivant et d’obtenir de nouveaux aperçus de la machinerie cellulaire. En collaboration avec des chercheurs de l’université suédoise de Lund, il est maintenant possible de transposer le phénomène du repliement des protéines sur un ordinateur quantique. Le quantificateur à ondes D JUPSI de l’infrastructure d’ordinateurs quantiques JUNIQ possède plus de 5000 qubits et est le premier appareil de cette taille en dehors des USA. Dans l’interview, Mohanty nous explique l’ intérêt de ce travail de pionnier.
En quoi consiste la tâche que vous vous êtes fixée, Dr Mohanty ?
Je dirais que ce qui compte vraiment, c’est que nous ayons démontré l’utilité pratique des ordinateurs quantiques dans notre domaine. Les ordinateurs quantiques sont en effet une technologie encore assez nouvelle. On ne sait pas encore très bien comment les programmer pour résoudre des problèmes scientifiques. C’est quelque chose de complètement différent du High-Performance Computing (HPC) classique avec des superordinateurs classiques.

Concrètement, nous avons étudié le repliement des protéines à l’aide d’un modèle très simple. Les protéines sont des éléments constitutifs importants de la vie. Elles remplissent une large palette de tâches. Parmi celles-ci figurent par exemple le transport de substances et la structure des cellules. Elles ne peuvent remplir toutes ces fonctions que si elles ont une forme bien précise, qu’elles atteignent grâce à un processus appelé repliement des protéines. L’une des nombreuses raisons du grand intérêt porté à ce processus est le lien entre les maladies neurodégénératives comme la maladie d’Alzheimer ou de Parkinson et le mauvais repliement des protéines. Nous espérons que les ordinateurs quantiques nous permettrons d’améliorer la compréhension de tels phénomènes.
Pourquoi la prédiction du repliement des protéines est-elle si exigeante en termes de calcul ?
Copyright : – Forschungszentrum Jülich / Ralf-Uwe Limbach
Les protéines sont de longues chaînes flexibles d’acides aminés. L’une de leurs propriétés est qu’elles s’agglomèrent spontanément en des formes tridimensionnelles très spécifiques lorsqu’on les place dans une solution, par exemple dans l’eau. Pour préfigurer cette forme, il faut connaître l’ordre des acides 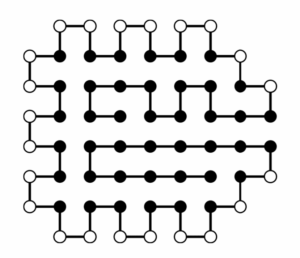 aminés qui composent la chaîne de protéines.
aminés qui composent la chaîne de protéines.
Exemple de la structure calculée d’une chaîne de 64 acides aminés avec l’énergie la plus faible connue. Les points remplis et entourés indiquent les éléments hydrophobes (H) et hydrosolubles/polaires (P). Copyright : – A. Irbäck et al., Phys. Rev. Research, DOI : 10.1103/PhysRevResearch.4.043013 (CC BY 4.0)
Lorsque l’on modélise ce processus de convolution sur ordinateur, il y a beaucoup d’essais à faire. On peut s’imaginer que c’est comme si l’on essayait de calculer toutes les différentes possibilités d’assemblage d’un collier pour trouver le « meilleur » arrangement. De plus, l’étude de chaque arrangement est très intensive en termes de calcul en raison du grand nombre de particules impliquées. Cela fait des millions d’interactions à prendre en compte.
Comment l’ordinateur quantique fonctionne-t-il par rapport à un superordinateur ou un ordinateur numérique classique ?
La tâche que nous avons résolue est, de par sa complexité, encore à des années des problèmes que nous résolvons normalement avec des supercalculateurs classiques. Avec ces derniers, il s’agit généralement de simulations à grande échelle et à l’échelle atomique. Sur l’analyseur quantique à ondes D, nous avons en revanche utilisé un modèle HP très réduit. Celui-ci simplifie considérablement le problème en ne conservant que les propriétés physiques essentielles du processus de convolution. Ce faisant, nous ignorons le milieu environnant. Les acides aminés sont divisés en seulement deux types et considérés, de manière simplifiée, comme des sphères occupant des positions dans une grille.
« L’étude de chaînes de 64 acides aminés avec un ordinateur quantique pose un problème difficile, même avec des modèles aussi simplifiés, le résultat est déjà très remarquable ». Sandipan Mohanty
Les simulations correspondantes peuvent également être réalisées de manière classique. Pour cela, un ordinateur portable suffit. Le temps nécessaire au calcul n’est pas très différent, dans les deux cas, il dure une à deux minutes. Toutefois, cette valeur n’a en fait aucune signification. La qualité des résultats est bien plus importante. Et sur ce point, le scanner quantique s’en sort nettement mieux. Il a été relativement facile d’obtenir sur JUPSI un taux de réussite de 100 % pour trouver les structures ayant la plus faible énergie. En revanche, avec des ordinateurs classiques, des simulations comparables pour une chaîne de 30 acides aminés n’atteignent que 80 pour cent. Pour les protéines plus complexes, composées de 48 ou 64 blocs d’acides aminés, les résultats sont encore nettement moins bons. Ici aussi, le quantificateur a toujours fourni le bon résultat.
Pourquoi l’ordinateur quantique est-il ici plus précis qu’un ordinateur classique ?
Parce qu’il profite de certains aspects de cette tâche scientifique. Le temps de calcul nécessaire pour prendre en compte toutes les conformations des protéines sur un ordinateur classique est astronomiquement élevé. Il croît de manière exponentielle avec la longueur de la chaîne protéique. Pour une chaîne de deux particules, il y a peut-être dix possibilités. Pour trois particules, il y en a déjà une centaine. Mais avec 100 particules – ce qui est encore très peu pour une protéine – il faudrait calculer des milliards de fois plus de variations qu’il n’y a d’atomes dans l’univers.
De nombreuses astuces sont utilisées pour pouvoir effectuer un calcul pertinent. Notre groupe au Jülich Supercomputing Centre et mes partenaires en Suède se sont spécialisés dans les simulations dites de Monte Carlo. Cette méthode est basée sur la physique statistique et l’échantillonnage stochastique. Bien que les simulations infiniment longues garantissent des solutions correctes, de brefs passages peuvent présenter des erreurs importantes. Dans la pratique, on essaie donc de réaliser les simulations « suffisamment longtemps » pour que les erreurs estimées soient moindres.
C’est là que réside l’avantage du scanner quantique. Cette machine, si elle est correctement programmée, peut effectuer cette approximation de manière très directe via ses couplages de mécanique quantique. En fait, il s’agit d’une sorte d’expérience physique compliquée qui résout automatiquement l’équation. Dans notre problème, cela a apparemment conduit à ce que des temps d’exécution relativement courts soient nécessaires pour obtenir de très bonnes réponses. Le fait que cela fonctionne si bien dans la pratique nous a en effet quelque peu surpris.
Quelles sont les perspectives d’application de l’informatique quantique ?
Notre travail n’est qu’une première étape. Les ordinateurs quantiques actuels n’ont que quelques qubits. Le système D-Wave en a déjà 5000, ce qui est beaucoup. Mais pour la plupart des problèmes de recherche intéressants, il faut davantage de qubits. Nous sommes encore loin des simulations réalisées dans la recherche sur les médicaments à l’aide des superordinateurs. Je pars du principe que nous devrons encore attendre deux ou trois générations d’appareils avant de pouvoir résoudre de tels problèmes avec un ordinateur quantique.
Mais je suis très optimiste. Car contrairement aux recherches précédentes, nous avons pu conserver la simplicité de notre formulation à mesure que la taille du système augmentait. Cela ouvre une voie potentiellement plus simple pour étudier des problèmes beaucoup plus complexes sur des ordinateurs quantiques.
Source : https://www.fz-juelich.de/de
Entretien : Tobias Schlößer
Contacts :
Publication original :
Anders Irbäck, Lucas Knuthson, Sandipan Mohanty, and Carsten Peterson
Folding lattice proteins with quantum annealing
Phys. Rev. Research (published 10 October 2022), DOI: 10.1103/PhysRevResearch.4.043013